

A spinal cord stimulator is an implantable medical device used to manage chronic pain, most often involving the back or spine. These systems are marketed as a way to reduce pain by interrupting nerve signals before they reach the brain. But for a growing number of patients, the device fails to help. It introduces new and sometimes permanent problems, including electrical shocks, burning pain, infections, lead migration, hardware failure, and repeat surgeries to reposition or remove equipment that was supposed to improve quality of life.
This page explains spinal cord stimulator lawsuits and why they are being filed nationwide. It focuses on what patients are alleging, how these devices have failed in real-world use, and why many of these cases go beyond ordinary medical malpractice claims. The most serious lawsuits do not center on a single surgical mistake. They examine how modern spinal cord stimulators were designed, tested, and approved, and whether patients were ever adequately warned about the risks that now recur repeatedly in medical records and FDA reports.
Many people arrive here with a practical question in mind: what do spinal cord stimulator settlement amounts look like, and how does compensation get calculated when a device causes lasting harm? That question cannot be answered in isolation. Settlement amounts and payouts are driven by the full medical timeline, including the costs of repeat surgeries, explantation, and permanent loss of function, as well as the downstream consequences when a pain-management device leaves someone worse off than before it was implanted.
What follows is a detailed breakdown of the allegations, the regulatory history behind these devices, the types of injuries being reported, and how spinal cord stimulator compensation is evaluated in serious cases.
If you or a loved one suffered complications after implantation, this page is meant to help you understand not just whether you may have a claim, but why these cases are being taken seriously across the country.
Get a free no-obligation consultation or call us today at 800-553-8082.
Spinal Cord Stimulator Lawsuit Updates
The spinal cord stimulator litigation is new, fast-moving, and constantly changing as more lawsuits are filed, more medical device issues come into focus, and courts begin deciding how these cases should be organized.
Our lawyers are following these developments closely and will continue updating this page with the latest filings, court rulings, MDL activity, trial developments, and settlement news as the litigation moves forward.
July 28, 2026
New Mexico Man Alleges Abbott Spinal Cord Stimulator Caused Shocks and Neurologic Injuries
A new lawsuit filed by an Alamogordo, New Mexico man alleges that an Abbott Proclaim XR5 spinal cord stimulator malfunctioned after implantation, causing painful electrical shocks, increased pain, leg weakness, urinary and fecal incontinence, and lasting neurologic injuries.
The plaintiff says a Medtronic spinal cord stimulator had controlled his chronic pain for approximately five years before his medical providers recommended replacing it with an Abbott device. According to the complaint, an Abbott representative described the Proclaim XR5 as safe, clinically validated, MRI compatible, superior to the prior system, capable of providing long-term relief, and easy to remove. The representative allegedly encouraged him to proceed directly to permanent implantation without first completing a trial period.
The device was implanted in Alamogordo in July 2023. The lawsuit alleges that Abbott failed to disclose that it had recently initiated a recall of Proclaim devices that could become unable to exit MRI mode. The plaintiff says he was not told about the recall until October 2023.
After the surgery, he allegedly developed profound leg weakness and lost control of his bladder and bowels. Abbott representatives reportedly told him that the symptoms were part of the healing process and repeatedly reprogrammed the stimulator without meaningful physician supervision. Despite several visits to the Las Cruces Pain Center, the plaintiff says the device increased his pain rather than relieving it.
In February 2024, an MRI technician allegedly attempted to place the stimulator into MRI mode, causing it to deliver a violent electric shock. Neither the technician nor an Abbott representative could successfully activate MRI mode. The plaintiff later requested removal but says he struggled to find a physician willing to perform the procedure despite earlier assurances that explantation would be straightforward.
The Proclaim XR5 was surgically removed in February 2025 because of mechanical and therapeutic failure. The plaintiff alleges that he continues to experience chronic pain and an inability to control his bladder.
July 18, 2026
Boston Scientific MDL Initial Conference Set for August 5
Plaintiffs’ lawyers will meet by July 24 and propose a leadership structure for the Boston Scientific spinal cord stimulator MDL. Applications for lead counsel and liaison counsel are also due that day, along with a joint preliminary report addressing the early organization of the litigation.
Judge Josephine Staton will hold the first scheduling conference on August 5, 2026. The court is expected to address plaintiffs’ leadership appointments, pending motions, early case management issues, and the schedule for future status conferences. These are the first organizational steps in a new MDL.
As discussed in the updates below, separate requests are pending to consolidate spinal cord stimulator lawsuits against Abbott and Nevro into their own manufacturer-specific MDLs. If approved, those cases will proceed alongside the Boston Scientific litigation through coordinated discovery, pretrial rulings, and eventual bellwether trials.
July 14, 2026
Medtronic Wins Broad Dismissal But Manufacturing Claims Survive
U.S. District Judge Patrick J. Schiltz in Minnesota dismissed most of the claims brought by four patients who alleged that Medtronic used hundreds of FDA supplements to transform its spinal cord stimulators without submitting the redesigned devices to a new premarket approval process.
The court dismissed all claims brought by three plaintiffs because they waited too long to file under the applicable statutes of limitations. The judge also dismissed the plaintiffs’ Administrative Procedure Act claim against the FDA, finding that they lacked standing and had not identified a law requiring Medtronic to submit an entirely new premarket approval application rather than use the supplemental approval process.
This is a meaningful win for Medtronic because the ruling weakens the broad theory that the company improperly avoided FDA review by making hundreds of incremental changes to an older approved device. But the ruling is not fatal to the litigation.
The court allowed manufacturing defect claims from one plaintiff to proceed based on allegations that his 2024 spinal cord stimulator was not built according to Medtronic’s own design specifications. A related negligence claim also survived to the extent it alleges that Medtronic violated federal current good manufacturing practices.
The judge did not find that Medtronic spinal cord stimulators are free from defects or incapable of causing injury. Most of the dismissed claims failed because of filing deadlines, standing problems, or the way the regulatory theory was pleaded. Plaintiffs with timely claims and evidence that a device departed from Medtronic’s specifications or violated federal manufacturing requirements may still have viable cases.
June 5, 2026
Boston Scientific Spinal Cord Stimulator MDL Created
The federal court system has created a new MDL for Boston Scientific spinal cord stimulator lawsuits, but the consolidation is limited to Boston Scientific devices. Claims involving Abbott, Medtronic, and Nevro stimulators were left out for now.
The lawsuits allege that Boston Scientific spinal cord stimulators can fail to provide pain relief, deliver painful electric shocks, migrate, malfunction, or require removal surgery. The JPML found that the Boston Scientific cases share enough common issues to justify putting them before one judge in the Central District of California.
The panel declined to create a broader industry-wide MDL because the cases against Abbott, Medtronic, and Nevro involve different products, different regulatory histories, and no allegation that the manufacturers acted together. The Abbott cases were also not centralized because the pending cases were already in the same federal district, although the JPML left open the possibility of future coordination if those lawsuits grow.
June 1, 2026
Plaintiffs Seek Separate Abbott Spinal Cord Stimulator MDL
Plaintiffs are asking the U.S. Judicial Panel on Multidistrict Litigation to create a separate Abbott spinal cord stimulator MDL after the panel declined to form one industry-wide proceeding covering every manufacturer. The motion seeks centralization in the Central District of California, where the Boston Scientific cases are already pending.
The Abbott motion alleges common problems involving battery failures, shocks, burns, lead migration, device migration, nerve pain, and revision or explant surgery. If the panel grants centralization, the Abbott claims will remain individual lawsuits while sharing coordinated discovery, pretrial rulings, and potentially bellwether trials.
A separate manufacturer-specific MDL could give plaintiffs access to centralized discovery concerning Abbott’s design changes, warning decisions, adverse-event reporting, and response to recurring device failures. The panel has not yet decided the request.
What Will Spinal Stimulator Settlement Amounts Be?
When people ask about spinal stimulator settlement amounts, they are usually not asking out of curiosity. They are trying to understand whether the harm they suffered is being taken seriously and whether the legal system recognizes the scope of what went wrong. The truth is that spinal cord stimulator lawsuits are not really about chasing a number. They are intended to compensate for a cascade of losses that often follow implantation when these devices fail.
For many patients, the first injury is not the worst one. What follows can be months or years of repeat surgeries, hospitalizations, imaging studies, and failed attempts to fix a device that was supposed to reduce pain, not multiply it. Each revision surgery carries its own risks, costs, and recovery time. For some, the device never works as promised again. For others, it must be removed entirely, leaving them with permanent damage and no pain relief at all.
There is also a less visible cost that shows up in almost every serious case: loss of trust in the medical system. Patients often describe feeling dismissed when they report shocks, burning sensations, or worsening pain. They are told the symptoms are rare, expected, or unrelated, even as their quality of life steadily declines. That erosion of trust, combined with physical suffering and financial strain, is a core driver of these lawsuits.
Only after those realities are understood does it make sense to talk about settlement compensation and payouts. Higher spinal stimulator settlement amounts tend to be associated with cases involving clear, documented harm rather than temporary discomfort. The strongest cases often include multiple revision surgeries, complete explantation of the device, or injuries that permanently alter neurological function.
Illustrative Settlement Payout Scenarios
There is no reliable average settlement for spinal cord stimulator lawsuits at this stage. The estimates below reflect our lawyers’ experience and judgment about how injury severity may affect potential value if liability, causation, insurance, and collectability are established. They are not reported settlement averages, promises, or predictions of what any particular claimant will receive.
| Scenario | Clinical Evidence | Our Illustrative Estimate |
|---|---|---|
| One revision surgery | Documented shocks or worsening pain, one corrective procedure, and limited permanency. | $90,000 to $250,000 |
| Explantation required | Removal surgery with ongoing pain, scarring, or functional limitations. | $150,000 to $550,000 |
| Multiple surgeries or migration | Two or more revisions with documented migration, lead failure, or repeated loss of therapy. | $250,000 to $1,100,000 |
| Permanent neurological injury | Objective nerve damage, weakness, gait changes, paralysis, or another lasting neurological deficit. | $1,100,000 to $1,900,000 |
| Bowel, bladder, or autonomic injury | Permanent bowel or bladder loss, autonomic complications, and substantial loss of independence. | $1,900,000 to $2,500,000 |
What Will Drive Spinal Stimulator Compensation Payouts?
- Number of surgeries, including revision and explantation
- Objective proof of migration, lead failure, overheating, or malfunction
- Neurologic injury or bowel or autonomic dysfunction
- Medical expenses, wage loss, and diminished earning capacity
- Permanency, credibility, and the jurisdiction where the case is filed
Are Quick Settlements Possible?
You should not expect a quick settlement, although an individual case can resolve early. A lawsuit filed on October 17, 2025, alleged that a woman suffered severe and worsening injuries after receiving a spinal cord stimulator manufactured by Boston Scientific, claiming the device migrated, delivered painful electrical shocks, and failed to perform as promised. The complaint accused the company of selling a defective medical device and failing to warn patients and doctors about known risks, while also raising broader concerns about regulatory oversight.
The plaintiff alleged that the stimulator differed materially from the version originally approved by the FDA and that those changes increased the risk of harm without adequate testing or disclosure. The case fit into a growing wave of spinal cord stimulator litigation focusing less on surgical error and more on device design, post-approval modifications, and inadequate warnings.
The case was reportedly resolved in December 2025, less than 60 days after filing and before substantial pretrial discovery. The amount and terms were not disclosed. One early resolution does not establish a timetable or settlement value for other spinal cord stimulator claims.
Spinal Cord Stimulators
Spinal cord stimulation devices are Class III implantable neuromodulation systems intended to deliver controlled electrical impulses to the spinal cord to manage chronic, intractable pain. These systems generally include an implantable pulse generator, one or more electrical leads placed near the spinal cord, and an external controller that allows the patient to adjust stimulation levels.
The theory behind spinal cord stimulation is that targeted electrical impulses applied to the dorsal columns of the spinal cord can interfere with or alter the transmission of pain signals before they reach the brain. By modulating these signals, the devices are intended to provide relief for patients whose pain has not responded to conventional treatments.
Despite their intended benefits, spinal cord stimulation systems carry significant and well-documented risks. Reported complications include device migration, lead fracture or displacement, battery malfunction, infection, neurological injury related to stimulation, worsening pain, and autonomic dysfunction.
Because of these substantial risks, the FDA classifies spinal cord stimulation systems as Class III medical devices. This designation requires rigorous Premarket Approval and supplemental review for any design or functional modifications that could affect the device’s safety or effectiveness.
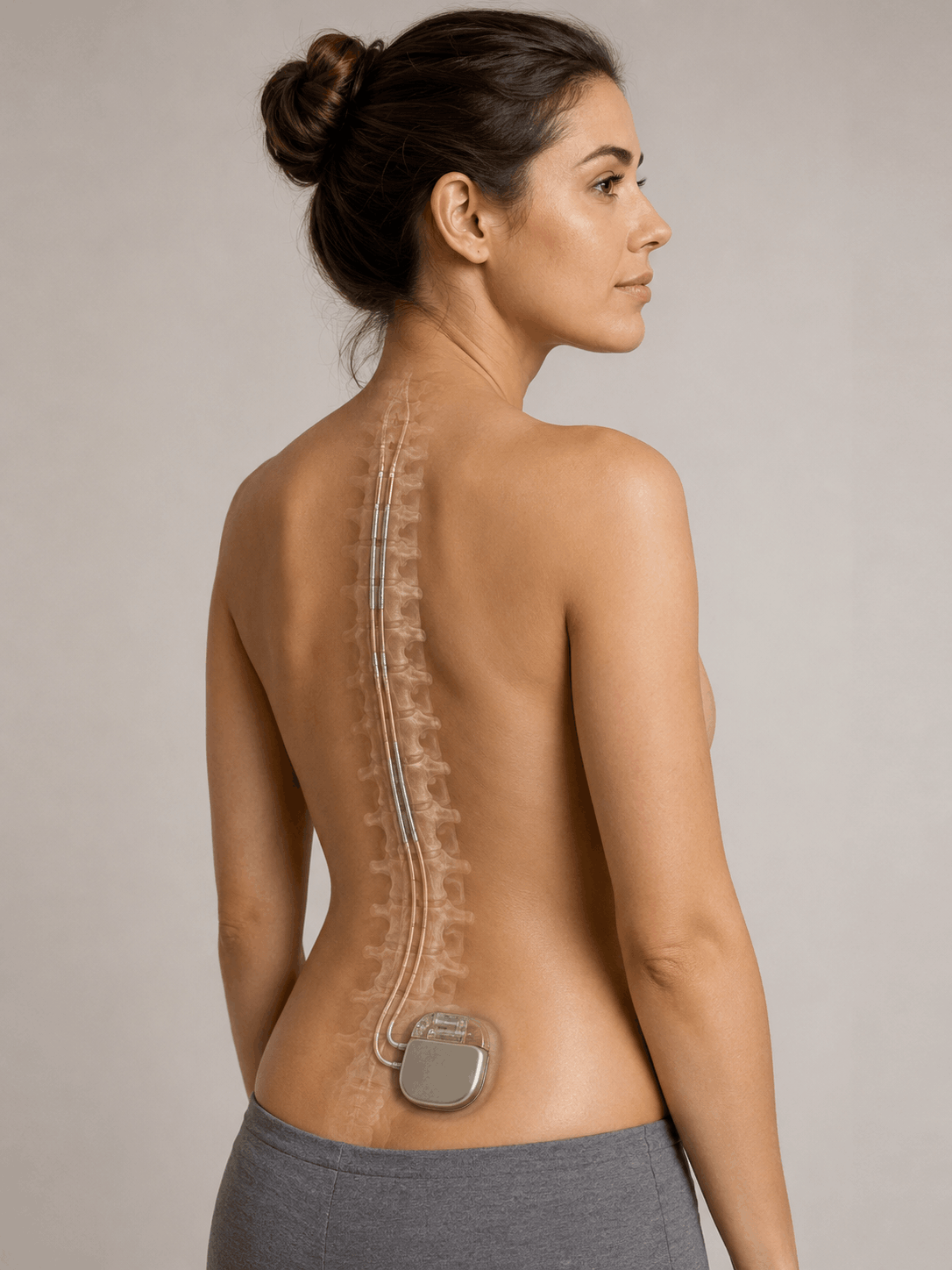
Spinal Cord Stimulator Lawsuits: Allegations of Defective Medical Devices
Across the United States, patients are filing lawsuits involving spinal cord stimulators (SCS), alleging these implantable pain-management devices were defectively designed and unreasonably dangerous. According to FDA adverse-event data, thousands of complaints have been reported involving spinal cord stimulators, including burns, infections, device migration, electrical malfunction, and the need for repeated corrective surgeries.
Major manufacturers—including Boston Scientific, Medtronic, Abbott, and Nevro Corp—are accused of failing to adequately warn patients and physicians about known risks associated with these devices. Independent scientific studies have raised concerns that spinal cord stimulators are prone to lead migration, hardware failure, and electrical defects, which can result in serious neurological injury and long-term complications.
Many patients report they were never fully informed of these dangers before implantation. After suffering burns, infections, worsening pain, paralysis, or undergoing revision surgeries or complete device removal, patients often face significant physical pain, emotional distress, and financial hardship. As a result, injured individuals and their families are pursuing spinal cord stimulator lawsuits to seek compensation and accountability.
What separates spinal cord stimulator lawsuits from ordinary medical malpractice cases is not just the severity of the injuries, but the regulatory and design history behind these devices. Many of the most serious claims now being filed do not focus solely on surgical error or isolated malfunction. Instead, they examine how modern spinal cord stimulators were approved, how dramatically they changed over time, and whether patients were ever protected by the safety testing they were led to believe occurred.
Reported Spinal Cord Stimulator Complications
Spinal cord stimulator injuries being alleged in lawsuits include:
- Lead migration requiring revision surgery
- Electrical malfunction causing shocks or burns
- Device overheating
- Hardware failure
- Infection
- Scar tissue complications
- Worsening neuropathic pain
- Paralysis or weakness
- Autonomic dysfunction
- Bowel or bladder loss
- Cardiac complications
- Death following postoperative complications
One reported case involved an elderly patient, whom we talk about below, who developed Ogilvie syndrome following implantation. The complication progressed to bowel rupture and multi-organ failure. While causation remains disputed, similar autonomic complications have been reported following neuromodulation procedures.
Another lawsuit alleges a WaveWriter Alpha stimulator migrated and delivered painful shocks, with company representatives allegedly adjusting the device without a physician present.
These fact patterns are not identical. But the themes are consistent: device movement, electrical malfunction, worsening symptoms, and inadequate warning.
How FDA Approval Is Being Used as a Shield
One of the first things spinal cord stimulator manufacturers point to when patients are injured is FDA approval. The implication is simple and powerful: the device was approved, therefore it must be safe. But that framing omits how spinal cord stimulators reached the market and how they have evolved.
Spinal cord stimulators are classified as Class III medical devices, the highest risk category under federal law. So in theory, that classification means the device must undergo rigorous premarket approval, including clinical testing, before it can be sold. That is the level of review patients reasonably expect. In practice, many of today’s spinal cord stimulators trace their approval back to much older devices, approved decades ago, through a regulatory pathway that allows manufacturers to make changes without submitting new clinical evidence each time.
Manufacturers are permitted to update devices through PMA supplements. These are intended to cover limited, incremental changes that do not meaningfully alter how a device works or how risky it is. The problem arises when companies rely on this process to roll out sweeping design changes over many years without ever returning to the FDA for a full review.
In the spinal cord stimulator space, manufacturers have introduced major modifications to battery systems, lead designs, stimulation waveforms, wireless communication features, posture-adaptive algorithms, and multi-source current delivery. These are not cosmetic tweaks. They directly affect how electrical signals are delivered to the spinal cord and how the device behaves inside the body.
Yet many of these changes were cleared through paperwork rather than new human trials. As a result, patients may be implanted with devices that are materially different from the versions originally approved, even though the FDA approval label remains the same. From the patient’s perspective, the device appears fully vetted. From a regulatory standpoint, the safety data often lags far behind the technology.
This regulatory gap matters in real-world terms. When a device malfunctions, migrates, overheats, or delivers unintended electrical shocks, manufacturers often argue that these outcomes fall within “known risks.” But a risk cannot be meaningfully known if the device has evolved beyond the version that was ever properly studied.
Lawsuits now being filed across the country allege that manufacturers exploited this regulatory structure by stacking change upon change without conducting the testing needed to reveal long-term failure rates or rare but devastating complications. Plaintiffs also allege that warnings never reflected how these newer systems behaved once implanted in patients.
The result is a familiar pattern in modern medical device litigation. FDA approval becomes a talking point, not a safeguard. Devices reach patients faster than the science can keep up, and serious complications are only fully recognized after thousands of people have already been exposed.
This is why many spinal cord stimulator lawsuits focus not just on individual injuries but also on whether manufacturers complied with the spirit and intent of the regulatory system. The core allegation is not that innovation is bad. It is that innovation without accountability shifts the risk onto patients who never agreed to be test subjects.
Virginia Woman Sues FDA and Boston Scientific Over Defective Device
A Virginia woman has filed a spinal cord stimulator lawsuit against Boston Scientific and the U.S. Food and Drug Administration, alleging that a defective spinal cord stimulator caused severe and worsening injuries after implantation.
According to the complaint, the woman received a WaveWriter Alpha spinal cord stimulator to manage chronic pain, but instead, it made things worse, and she experienced electrical shocks, device migration, and escalating physical harm.
The lawsuit further alleges that the device moved from its intended position and delivered painful electrical impulses that were not only ineffective but also dangerous. Doctors sometimes call this migration, and it can cause real damage to the patient in many different contexts.
The patient also claims that company representatives adjusted and altered the device without her physician present, dismissing her concerns even as her condition deteriorated. If you are following this medical device, you would not be surprised, as the allegations mirror reports documented in FDA adverse-event data, in which other patients have described similar injuries linked to the same device system.
In addition to claims against the manufacturer, the lawsuit raises serious questions about regulatory oversight. The plaintiff alleges that Boston Scientific continued to market and sell a spinal cord stimulator that materially differed from the version originally approved by the FDA, without undergoing proper supplemental review or safety testing. The complaint asserts that these changes increased the risk of injury while depriving patients and physicians of critical information needed to make informed decisions.
The case highlights a growing concern in spinal cord stimulator litigation: that device manufacturers may prioritize speed to market and continued sales over patient safety, while regulators fail to intervene despite mounting evidence of harm. Although the case remains pending, it reflects broader allegations being raised nationwide by patients who say they were never adequately warned about the risks associated with these devices—and who now face lasting pain, repeated procedures, and diminished quality of life.
When Device Representatives Cross the Line Into Treatment
One of the more serious issues emerging in spinal cord stimulator litigation is that some lawsuits are not just about the device itself. They focus on the role manufacturer representatives play before and after implantation. The claim is that these representatives are not merely providing technical support. In some cases, they are accused of helping sell the implant by making assurances about long-term pain relief, clinical validation, and expected performance, then remaining heavily involved once problems begin.
A spinal cord stimulator lawsuit may involve more than a hardware malfunction. It may also examine whether sales-driven assurances influenced the decision to implant the device and whether repeated reprogramming delayed an independent medical assessment that the system was failing.
Plaintiffs allege that manufacturer representatives took an active role in programming and reprogramming spinal cord stimulators after implantation, sometimes across multiple visits and with limited physician involvement. This can blur the line between technical support and treatment, especially when the representative becomes the patient’s primary contact after pain worsens, stimulation becomes erratic, or the device stops providing relief.
These allegations become even more troubling when paired with claims of lead migration, electrical shocks, or sudden loss of therapeutic benefit. One of the recurring themes in the spinal stimulator lawsuits is that repeated device adjustments may be used as a temporary response to serious complications, even when the underlying issue may be hardware movement, system instability, or loss of efficacy that reprogramming cannot fix. This looks less like troubleshooting and more like a delay.
This strengthens several types of claims at once. It can support allegations involving negligent misrepresentation, breach of warranty, failure to warn, and broader negligence theories tied to post-implant conduct. It also gives juries a more concrete story. Instead of hearing only that a device failed, they hear that the manufacturer may have remained deeply involved while the patient’s condition worsened.
Abbott Recall History and Warning Failures
Another issue likely to matter in spinal cord stimulator litigation is whether patients and doctors were ever given a fair warning about how newer Abbott systems actually performed in the real world. The Abbott spinal cord stimulator lawsuits being filed do not just allege that these devices could fail in a general sense. We allege that important risks were understated, poorly communicated, or buried beneath broad assurances about safety and effectiveness.
Spinal cord stimulator complications are not limited to disappointing results. Painful electrical shocks, sudden loss of therapy, charging failures, communication problems between device components, lead migration, and worsening pain are all things patients have reported after implantation. In a device intended to treat chronic pain, those are not minor side effects. They go to the core question of whether the product was functioning safely at all.
Abbott also draws added scrutiny because of its recall history involving serious device problems. FDA Class I recalls are the agency’s most serious recall category, and recent complaints point to Proclaim-related recalls involving issues such as unintended painful stimulation, device shutdowns, and failure to deliver therapy. Plaintiffs argue that a recall history like this undercuts any suggestion that the reported problems are isolated or speculative.
The broader allegation is that the warnings never kept pace with the technology. As spinal cord stimulator systems evolved through repeated supplemental changes to firmware, battery architecture, wireless programming, and stimulation features, our contention is that the risks evolved too. But, as our lawsuits argue, patients were still not clearly told that the device being implanted may have been materially different from the version originally approved years earlier.
That warning gap is a big deal in these cases. A manufacturer does not get much mileage out of saying a risk was technically known if the actual device changed over time and the real-world complications became more serious, more frequent, or simply different from what patients and doctors were led to expect. That is why failure-to-warn claims remain central to this litigation, even with the preemption fight hanging over these cases.
Repeated Reprogramming After Implantation Can Be a Warning Sign
One issue that does not get enough attention in spinal cord stimulator lawsuits is what happens after the permanent device is implanted and the patient starts complaining that something is wrong.
Many patients are told that the answer is simple: the device just needs to be reprogrammed. A representative adjusts the settings. The patient is told to give it more time. Then the pain comes back, the stimulation becomes uncomfortable, the device stops helping, or new symptoms develop. The cycle repeats.
That pattern matters. Reprogramming is not automatically improper. These devices often require adjustments after implantation. But repeated reprogramming can become a red flag when it is used as the default answer to every complaint, especially when the patient is reporting shocks, numbness, worsening pain, loss of balance, falls, bladder or bowel symptoms, or loss of therapeutic benefit.
At some point, the question should shift from “Can we adjust the settings again?” to “Is the device failing?”
This is especially important when company representatives become the main people managing the device after surgery. Patients may reasonably believe the representative is acting like part of the medical team. But manufacturer representatives are not neutral treating physicians. They work for the very company that sold the device. So when they reassure patients that complications can be fixed with another programming change, that can delay a real medical evaluation of whether the stimulator has migrated, malfunctioned, lost efficacy, or should be removed.
In spinal cord stimulator lawsuits, this reprogramming cycle can become key evidence. In some cases, it will show that the device was not performing as represented. It also may show that warnings were far from adequate. An extremely strong allegation would be if the manufacturer remained involved after implantation in a way that shaped the patient’s treatment decisions, not just the device’s technical settings.
So if repeated reprogramming does not restore meaningful relief, or if symptoms are getting worse, the problem may not be a settings problem. It may be a device problem.
Patients who experience these problems should document each reprogramming visit, who performed it, whether a physician was present, what symptoms were reported, what settings were changed, and whether the change actually helped. Those details can matter later if the device must be revised, removed, or becomes the basis for a legal claim.
Why Medtronic Is Now Being Sued in Minnesota State Court
Most spinal cord stimulator lawsuits are filed in federal court or in the state where the patient was implanted and injured. But plaintiffs across the country are choosing to file Medtronic stimulator lawsuits in Minnesota state court.
Why? Medtronic is headquartered in Minneapolis. Its regulatory strategy, labeling decisions, and post-market conduct were directed from that location. Filing in Minnesota puts the case before a local court that examines the conduct of a local company, and it may help plaintiffs avoid transfer into federal MDL proceedings. It also means discovery into Medtronic’s internal decision-making, including how it evaluated the risks of successive hardware modifications and what it chose to communicate to the medical community, takes place closer to where those decisions were made.
For patients implanted in other states, this approach requires a choice: file at home, where the injury occurred, or file in Minnesota, where Medtronic is based and where many of the company’s key decisions were allegedly made. Neither approach is universally right.
Why the Trial Stimulator and Permanent Implant May Not Match
This is one of the most frustrating patterns we see in spinal cord stimulator cases, and one that juries will understand immediately.
The patient does the trial. The trial works. The pain drops. For the first time in months or years, there is real relief. The doctor says the numbers look good. Everyone agrees it is time to move forward with the permanent implant.
Then the permanent device goes in. And somewhere down the line, weeks, months, sometimes longer, the relief fades. The pain returns. The hardware shifts. The system fails. The patient is left with a device inside their body that no longer functions as intended.
The essence of the problem is that the trial was never designed to predict long-term performance. It was designed to get the patient to say yes. That is not an accident. Device manufacturers know that the trial period is the moment of decision. If the trial goes well, the patient moves forward. If it does not, they walk away. So the trial is engineered to succeed. Short duration. Controlled conditions. Maximum placebo effect. What it does not do is tell the patient whether the permanent implant will actually work six months or two years down the road.
Lawsuits now being filed challenge exactly this. We allege that the permanent device does not deliver the same results as the trial because the implanted system behaves differently over time, because the leads migrate or the hardware fails, or because the short-term trial window was never an honest preview of what permanent implantation would look like.
This matters because it reframes the entire case. The issue is not always that the patient got zero benefit. The issue is that the trial created false confidence in a treatment that the permanent device could not actually deliver. The patient did everything right. Followed the process and trusted the system. And ended up with the worst of both worlds. An invasive implant inside their body, and none of the long-term relief they were promised.
Our lawyers believe juries will understand why a successful temporary trial can influence a patient to accept permanent implantation. The legal question is whether the trial and marketing created reasonable expectations that the permanent system could not meet, and whether the patient received a fair warning about the risk of long-term failure, migration, and loss of efficacy.
Bringing a Spinal Cord Stimulator Lawsuit
If you or a loved one experienced complications after spinal cord stimulator implantation, including electrocution, burns, infections, neurological injury, or repeated surgeries, you may have legal options. Lawsuits seek compensation for medical costs, lost income, pain and suffering, and long-term disability, while holding manufacturers accountable for unsafe products and inadequate warnings.
Eligibility for a Spinal Cord Stimulator Lawsuit
Patients who received a spinal cord stimulator expecting relief, but instead experienced serious complications, may have grounds to pursue a legal claim. The cases lawyers like us are looking for involve more than just a bad outcome or an unfortunate side effect. They raise broader questions about how these devices were designed, tested, marketed, and regulated in the first place.
You may qualify if you or a loved one had a spinal cord stimulator implanted and later suffered injuries such as infection, shocks, burns, device migration, neurological damage, bowel, or autonomic complications. You have the most viable claims. The upper-tier cases for settlement would involve victims who need repeated revision surgeries or complete device removal. In many cases, patients report they were never clearly warned that these risks were possible, let alone likely.
Eligibility does not depend on whether the device is still implanted, whether symptoms appeared immediately, or whether the manufacturer labeled the complication as “rare.” What matters is whether the device failed to perform as promised—or introduced dangers that were not adequately disclosed. Lawsuits often focus on whether manufacturers minimized known risks, overstated benefits, or continued selling devices that had changed in meaningful ways without proper safety review.
Families may also have claims in cases involving severe disability or death following implantation, particularly where complications cascaded rapidly and left little opportunity for intervention. These cases are not just about individual harm, but about accountability within a system that often favors speed, scale, and market share over long-term patient safety.
If you believe your injury was dismissed, normalized, or treated as an acceptable tradeoff, you are not alone. Many of the claims now moving through courts nationwide began the same way, with patients realizing too late that the full story was never told.
Spinal Cord Stimulator Lawsuit FAQs
Is there a spinal cord stimulator MDL?
Yes. The JPML created MDL No. 3181 for Boston Scientific spinal cord stimulator product liability cases on June 5, 2026. The cases are centralized in the Central District of California before Judge Josephine L. Staton. Claims against Abbott, Medtronic, and Nevro are not part of that MDL, although separate centralization requests may be considered.
What are estimated spinal cord stimulator settlement amounts?
There is no established average settlement or payment schedule. Our illustrative estimates range from lower six figures for a documented revision case to more than $1 million for permanent neurological or autonomic injury. Those figures reflect our lawyers’ experience and judgment and are not reported averages or guaranteed results.
Which spinal cord stimulator manufacturers are being sued?
Current lawsuits involve devices made by Boston Scientific, Abbott, Medtronic, and Nevro. The legal and regulatory issues differ by manufacturer and device family, so the company name and exact model should be confirmed from the implant card or medical records.
Does explant surgery strengthen a spinal cord stimulator lawsuit?
It can. Explantation provides objective evidence that conservative measures and reprogramming did not solve the problem. Case value still depends on why the device was removed, whether a product defect caused the injury, the number of procedures, and the patient’s lasting symptoms.
Can I sue if the permanent stimulator never provided pain relief?
Lack of pain relief alone may not prove a product liability claim. The case becomes stronger when loss of efficacy is accompanied by lead migration, hardware failure, painful stimulation, misleading representations, repeated unsuccessful reprogramming, revision surgery, or explantation.
Does FDA approval prevent a spinal cord stimulator lawsuit?
FDA premarket approval creates a serious federal preemption defense, but it does not automatically bar every lawsuit. Some claims may proceed when they are based on a violation of federal requirements or a device that departed from its approved manufacturing specifications. The available theories depend heavily on the jurisdiction and the facts.
What records should I preserve?
Preserve the implant card, operative reports, imaging, revision and explant records, programming logs, communications with manufacturer representatives, photographs, symptom notes, medical bills, wage records, and the device itself if it is removed and can be retained through proper evidence procedures.
Contact Us About Spinal Cord Stimulator Lawsuits
Our firm is seeking spinal cord stimulator lawsuits nationwide. If you have suffered physical or mental harm as a result of a spinal cord stimulator, contact us today for a free case evaluation. Call us at 800-553-8082 or contact us online.
Page written by Ronald V. Miller Jr. This page is for patients and families trying to understand the connection between spinal cord stimulators and a variety of injuries, the federal MDLs, and potential civil compensation claims.